НИЈМЕГЕНОВ СИНДРОМ НА РАСКИНУВАЊЕ/ НИЈМЕГЕНОВ СИНДРОМ НА КРШЕЊЕ/ NIJMEGEN BREAKAGE SYNDROME

MKБ-10 Q87.8 (Q90.9, D81.9)
ПРЕВАЛЕНЦА – непозната, иако постојат податоци дека преваленцата кај новороденчињата со источнословенско потекло (Белорусија, Украина, Русија и Латвија) била проценета на 1/1 000 000 живородени, додека преваленцата во западнословенскиот регион (Полска, Словачка и Чешка) се проценува на 1/330 000.
ВОВЕД
Нијмегенов синдром на раскинување (НСР) е редок синдром на генетска, хромозомска нестабилност која најверојатно настанува како резултат на дефект во механизмот за поправка на ДНК со двојна врска и/или механизмот за анелирање за поправка на прекинот на двојната врска во ДНК. Ова автосомно рецесивно нарушување за првпат било опишано кај холандски пациенти кои најверојатно биле потомци на полски или чешки граѓани кои емигрирале во Холандија за време на 1600-тите по 30-годишната војна бидејќи поголемиот дел од пациентите со НСР всушност живеат во Полска и Чешка. При раѓањето, новороденчињата се манифестираат со микроцефалија, дисморфни црти на лицето кои стануваат позабележителни со возраста, одложување на растот, повторливи синопулмонални инфекции и екстремно висока фреквенција на малигни тумори во раната возраст. Моделот на наследување е автосомно рецесивен.
ЕТИОЛОГИЈА
Нијмегеновиот синдром на кршење е предизвикан од мутации во генот NBN (8q21-q24), конкретно во рамките на егзоните 6-10, кои доведуваат до делумно функционални скратени фрагменти на нибрин, генски производ вклучен во поправка на прекините на ДНК со двојна врска. Над 90 % од пациентите од словенско потекло се хомозиготни.
СИМПТОМИ И ДИЈАГНОСТИЦИРАЊЕ
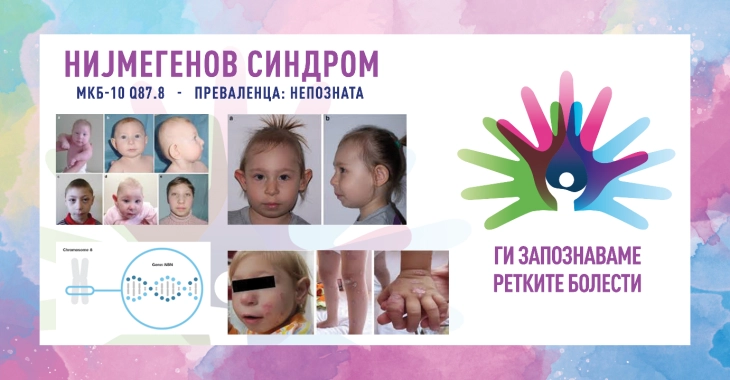
Клиничките манифестации не се типични и можат да се разликуваат според степенот на сериозност. Главните знаци се микроцефалија, присутна при раѓање која напредува со возраста, дисморфни црти на лицето (истакната средина нагласена со наведнато чело и повлекување на мандибулата), блага ретардација на растот и, кај женските пациенти, се јавува честа предвремена оваријална инсуфициенција. Пријавени се различни вродени аномалии, вклучително и во централен нервен систем (хидроцефалија, шизенцефалија, арахноидни цисти), респираторен тракт (расцеп на усна/непце, хоанална атрезија), урогенитален систем (потковица бубрег, ектопични/дистопични бубрези, хипоспадија, хипоспадија, крипторија), благи скелетни аномалии (пред- и постаксијална полидактилија, хипопластичен или двоен палец, клинодактилија на петтиот прст). Често се забележуваат дамки „café au lait“ и/или витилиго, а кај некои пациенти се јавуваат повеќе пигментирани нерви. Другите главни манифестации вклучуваат имунолошки дефицит со рекурентни инфекции на респираторниот тракт, силна предиспозиција за малигни тумори (претежно лимфоидни, но се појавуваат и цврсти тумори) и радиочувствителност. До 20-годишна возраст, над 40 % од пациентите развиваат малигно заболување и се склони кон развој на секундарни малигни тумори. Когнитивниот развој е блиску до просекот (нормален/граничен) во детството и предучилишната возраст, но интелектуалните вештини постепено опаѓаат со возраста (од благ до умерен).
Дијагнозата се заснова на клиничките манифестации и се потврдува или со секвенционирање на еден ген (обично за словенските популации) или со мултигенски панели за секвенционирање на следната генерација. Онаму каде што генетското тестирање е недостапно, дијагнозата може да биде поддржана со докази за хромозомска нестабилност (спонтана и индуцирана), зголемена клеточна чувствителност на јонизирачко зрачење, ин витро, комбинирана имунодефициенција и целосно отсуство на нибрин со целосна должина. Семејната историја (малигни заболувања, микроцефалија или хидроцефалија, рана смрт на брат или сестра) исто така може да ја дефинира дијагнозата. Особено важно е да се исклучат диференцијалните дијагнози, како што се Fanconi-анемија, LIG4-синдром, Cernunnos-XLF-дефицит, нарушување слично на NBS, нарушување слично на атаксија-телеангиектазија и Блумов синдром.
ПОСЛЕДИЦИ
Прогнозата е лоша, со малигнитетот како главна причина за смрт. Пациентите кои развиле лимфом или леукемија починале од прогресија на болеста, релапси и секундарни малигни тумори. Корисниот ефект на HSCT врз долгорочното преживување е потврден во неодамнешните студии.
ТРЕТМАН
За жал, и за овој синдром, како и повеќето, не постои специфична терапија. Од големо значење е раната дијагноза со цел да се избегнат тешки рекурентни инфекции, непотребна изложеност на зрачење за дијагностички цели и негативни ефекти од радиотерапијата на туморите. Родителите на засегнатото дете се задолжителни носители на мутациите на НСК и на тој начин, за секоја бременост, постои 25 % ризик за потомството да ја наследи болеста. На носителите на словенска основачка мутација треба да им се понуди следење на ракот, особено на дојката кај жените и на простатата кај мажите. На пациентите им е потребен мултидисциплинарен третман и долгорочно следење (малигнитет, имунодефициенција, раст, хипергонадотропен хипогонадизам кај жени). Следењето на имунолошкиот систем е исклучително важно во текот на животот; неопходни се специфични начини и типови на имунизација (се препорачуваат ацелуларни вакцини). Во случај на лимфоидна малигност, се препорачува трансплантација на хематопоетски матични клетки (HSCT) по првата целосна ремисија. Во поглед на припадничките на женскиот пол, се нуди следење на напредокот од 12-годишна возраст (ендокринолог/гинеколог) и хормонска терапија за соодветната возраст.

